HIV
20.52
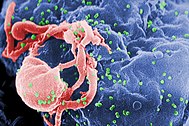
Human immunodeficiency virus (HIV) is a lentivirus (a member of the retrovirus family) that causes acquired immunodeficiency syndrome (AIDS),[1][2] a condition in humans in which the immune system begins to fail, leading to life-threatening opportunistic infections. Infection with HIV occurs by the transfer of blood, semen, vaginal fluid, pre-ejaculate, or breast milk. Within these bodily fluids, HIV is present as both free virus particles and virus within infected immune cells. The four major routes of transmission are unsafe sex, contaminated needles, breast milk, and transmission from an infected mother to her baby at birth (vertical transmission). Screening of blood products for HIV has largely eliminated transmission through blood transfusions or infected blood products in the developed world.
HIV infection in humans is considered pandemic by the World Health Organization (WHO). From its discovery in 1981 to 2006, AIDS killed more than 25 million people.[3] HIV infects about 0.6% of the world's population.[3] In 2005 alone, AIDS claimed an estimated 2.4–3.3 million lives, of which more than 570,000 were children. A third of these deaths are occurring in Sub-Saharan Africa, retarding economic growth and increasing poverty.[4] According to current estimates, HIV is set to infect 90 million people in Africa, resulting in a minimum estimate of 18 million orphans.[5] Antiretroviral treatment reduces both the mortality and the morbidity of HIV infection, but routine access to antiretroviral medication is not available in all countries.[6]
HIV infects primarily vital cells in the human immune system such as helper T cells (to be specific, CD4+ T cells), macrophages, and dendritic cells. HIV infection leads to low levels of CD4+ T cells through three main mechanisms: First, direct viral killing of infected cells; second, increased rates of apoptosis in infected cells; and third, killing of infected CD4+ T cells by CD8 cytotoxic lymphocytes that recognize infected cells. When CD4+ T cell numbers decline below a critical level, cell-mediated immunity is lost, and the body becomes progressively more susceptible to opportunistic infections.
Most people infected with HIV eventually develop AIDS. These individuals mostly die from opportunistic infections or malignancies associated with the progressive failure of the immune system.[7] HIV progresses to AIDS at a variable rate affected by viral, host, and environmental factors; HIV-specific treatment delays this process. Most will progress to AIDS within 10 years of HIV infection: some will have progressed much sooner, and some will take much longer.[8][9] Treatment with anti-retrovirals increases the life expectancy of people infected with HIV. Even after HIV has progressed to diagnosable AIDS, the average survival time with antiretroviral therapy was estimated to be more than 5 years as of 2005.[10] Without antiretroviral therapy, someone who has AIDS typically dies within a year.[11]
Classification
HIV is a member of the genus Lentivirus,[12] part of the family of Retroviridae.[13] Lentiviruses have many common morphologies and biological properties. Many species are infected by lentiviruses, which are characteristically responsible for long-duration illnesses with a long incubation period.[14] Lentiviruses are transmitted as single-stranded, positive-sense, enveloped RNA viruses. Upon entry of the target cell, the viral RNA genome is converted to double-stranded DNA by a virally encoded reverse transcriptase that is present in the virus particle. This viral DNA is then integrated into the cellular DNA by a virally encoded integrase, along with host cellular co-factors,[15] so that the genome can be transcribed. After the virus has infected the cell, two pathways are possible: either the virus becomes latent and the infected cell continues to function or the virus becomes active and replicates, and a large number of virus particles that can then infect other cells are liberated.
There are two species of HIV known to exist: HIV-1 and HIV-2. HIV-1 is the virus that was initially discovered and termed LAV. It is more virulent, more infective,[16] and is the cause of the majority of HIV infections globally. The lower infectivity of HIV-2 compared to HIV-1 implies that fewer of those exposed to HIV-2 will be infected per exposure. Because of its relatively poor capacity for transmission, HIV-2 is largely confined to West Africa.[17]
| Species | Virulence | Infectivity | Prevalence | Inferred origin |
|---|---|---|---|---|
| HIV-1 | High | High | Global | Common Chimpanzee |
| HIV-2 | Lower | Low | West Africa | Sooty Mangabey |
Signs and symptoms
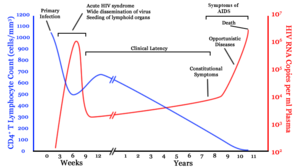
Infection with HIV-1 is associated with a progressive decrease of the CD4+ T cell count and an increase in viral load. The stage of infection can be determined by measuring the patient's CD4+ T cell count, and the level of HIV in the blood.
HIV infection has basically four stages: incubation period, acute infection, latency stage and AIDS. The initial incubation period upon infection is asymptomatic and usually lasts between two and four weeks. The second stage, acute infection, lasts an average of 28 days and can include symptoms such as fever, lymphadenopathy (swollen lymph nodes), pharyngitis (sore throat), rash, myalgia (muscle pain), malaise, and mouth and esophageal sores.
The latency stage, which occurs third, shows few or no symptoms and can last anywhere from two weeks to twenty years and beyond. AIDS, the fourth and final stage of HIV infection shows as symptoms of various opportunistic infections.
A study of French hospital patients found that approximately 0.5% of HIV-1 infected individuals retain high levels of CD4 T-cells and a low or clinically undetectable viral load without anti-retroviral treatment. These individuals are classified as HIV controllers or long-term nonprogressors.[18]
Acute HIV infection
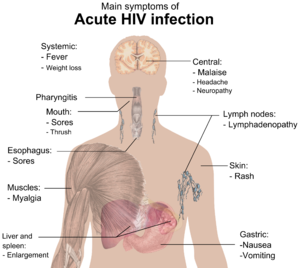
The initial infection with HIV generally occurs after transfer of body fluids from an infected person to an uninfected one. The first stage of infection, the primary, or acute infection, is a period of rapid viral replication that immediately follows the individual's exposure to HIV leading to an abundance of virus in the peripheral blood with levels of HIV commonly approaching several million viruses per mL.[19]
This response is accompanied by a marked drop in the numbers of circulating CD4+ T cells. This acute viremia is associated in virtually all patients with the activation of CD8+ T cells, which kill HIV-infected cells, and subsequently with antibody production, or seroconversion. The CD8+ T cell response is thought to be important in controlling virus levels, which peak and then decline, as the CD4+ T cell counts rebound to around 800 cells per µL (the normal blood value is 1200 cells per µL ). A good CD8+ T cell response has been linked to slower disease progression and a better prognosis, though it does not eliminate the virus.[20]
During this period (usually 2–4 weeks post-exposure) most individuals (80 to 90%) develop an influenza or mononucleosis-like illness called acute HIV infection, the most common symptoms of which may include fever, lymphadenopathy, pharyngitis, rash, myalgia, malaise, mouth and esophagal sores, and may also include, but less commonly, headache, nausea and vomiting, enlarged liver/spleen, weight loss, thrush, and neurological symptoms. Infected individuals may experience all, some, or none of these symptoms. The duration of symptoms varies, averaging 28 days and usually lasting at least a week.[21]
Because of the nonspecific nature of these symptoms, they are often not recognized as signs of HIV infection. Even if patients go to their doctors or a hospital, they will often be misdiagnosed as having one of the more common infectious diseases with the same symptoms. As a consequence, these primary symptoms are not used to diagnose HIV infection, as they do not develop in all cases and because many are caused by other more common diseases. However, recognizing the syndrome can be important because the patient is much more infectious during this period.[22]
Latency stage
A strong immune defense reduces the number of viral particles in the blood stream, marking the start of the infection's clinical latency stage. Clinical latency can vary between two weeks and 20 years. During this early phase of infection, HIV is active within lymphoid organs, where large amounts of virus become trapped in the follicular dendritic cells (FDC) network.[23]
The surrounding tissues that are rich in CD4+ T cells may also become infected, and viral particles accumulate both in infected cells and as free virus. Individuals who are in this phase are still infectious. During this time, CD4+ CD45RO+ T cells carry most of the proviral load.[24]
[edit] AIDS
- For more details on this topic, see AIDS Diagnosis, AIDS Symptoms and WHO Disease Staging System for HIV Infection and Disease
When CD4+ T cell numbers decline below a critical level of 200 cells per µL, cell-mediated immunity is lost, and infections with a variety of opportunistic microbes appear. The first symptoms often include moderate and unexplained weight loss, recurring respiratory tract infections (such as sinusitis, bronchitis, otitis media, pharyngitis), prostatitis, skin rashes, and oral ulcerations.
Common opportunistic infections and tumors, most of which are normally controlled by robust CD4+ T cell-mediated immunity then start to affect the patient. Typically, resistance is lost early on to oral Candida species and to Mycobacterium tuberculosis, which leads to an increased susceptibility to oral candidiasis (thrush) and tuberculosis. Later, reactivation of latent herpes viruses may cause worsening recurrences of herpes simplex eruptions, shingles, Epstein-Barr virus-induced B-cell lymphomas, or Kaposi's sarcoma.
Pneumonia caused by the fungus Pneumocystis jirovecii is common and often fatal. In the final stages of AIDS, infection with cytomegalovirus (another herpes virus) or Mycobacterium avium complex is more prominent. Not all patients with AIDS get all these infections or tumors, and there are other tumors and infections that are less prominent but still significant.
Pathophysiology
Transmission
| Exposure Route | Estimated infections per 10,000 exposures to an infected source | |||
|---|---|---|---|---|
| Blood Transfusion | 9,000[26] | |||
| Childbirth | 2,500[27] | |||
| Needle-sharing injection drug use | 67[28] | |||
| Percutaneous needle stick | 30[29] | |||
| Receptive anal intercourse* | 50[30][31] | |||
| Insertive anal intercourse* | 6.5[30][31] | |||
| Receptive penile-vaginal intercourse* | 10[30][31][32] | |||
| Insertive penile-vaginal intercourse* | 5[30][31] | |||
| Receptive oral intercourse*§ | 1†[31] | |||
| Insertive oral intercourse*§ | 0.5†[31] | |||
| * assuming no condom use § source refers to oral intercourse performed on a man | ||||
| † "best-guess estimate" | ||||
Three main transmission routes for HIV have been identified. HIV-2 is transmitted much less frequently by the mother-to-child and sexual route than HIV-1.
Sexual
The majority of HIV infections are acquired through unprotected sexual relations. Sexual transmission can occur when infected sexual secretions of one partner come into contact with the genital, oral, or rectal mucous membranes of another. In high-income countries, the risk of female-to-male transmission is 0.04% per act and male-to-female transmission is 0.08% per act. For various reasons, these rates are 4 to 10 times higher in low-income countries.[33]
The correct and consistent use of latex condoms reduces the risk of sexual transmission of HIV by about 85%.[34] However, spermicide may actually increase the transmission rate.[35][36][37]
A meta-analysis of 27 observational studies conducted prior to 1999 in sub-Saharan Africa indicated that male circumcision reduces the risk of HIV infection.[38] However, a subsequent review indicated that the correlation between circumcision and HIV in these observational studies may have been due to confounding factors.[39] In addition, concerns were raised about the potential for spread of HIV by unsterilized blades during ritual circumcision.[40] Later trials, in which uncircumcised men were randomly assigned to be medically circumcised in sterile conditions and given counseling and other men were not circumcised, have been conducted in South Africa,[41] Kenya,[42] and Uganda[43] showing reductions in female-to-male sexual HIV transmission of 60%, 53%, and 51% respectively. As a result, a panel of experts convened by WHO and the UNAIDS Secretariat has "recommended that male circumcision now be recognized as an additional important intervention to reduce the risk of heterosexually acquired HIV infection in men."[44]
Studies of HIV among women who have undergone female genital cutting (FGC) have reported mixed results; for details see Female genital cutting#HIV.
Blood or blood product
In general if infected blood comes into contact with any open wound, HIV may be transmitted. This transmission route can account for infections in intravenous drug users, hemophiliacs, and recipients of blood transfusions (though most transfusions are checked for HIV in the developed world) and blood products. It is also of concern for persons receiving medical care in regions where there is prevalent substandard hygiene in the use of injection equipment, such as the reuse of needles in Third World countries. Health care workers such as nurses, laboratory workers, and doctors have also been infected, although this occurs more rarely. Since transmission of HIV by blood became known medical personnel are required to protect themselves from contact with blood by the use of universal precautions. People who give and receive tattoos, piercings, and scarification procedures can also be at risk of infection.
HIV has been found at low concentrations in the saliva, tears and urine of infected individuals, but there are no recorded cases of infection by these secretions and the potential risk of transmission is negligible.[45] It is not possible for mosquitoes to transmit HIV.[46]
[edit] Mother-to-child
The transmission of the virus from the mother to the child can occur in utero (during pregnancy), intrapartum (at childbirth), or via breast feeding. In the absence of treatment, the transmission rate up to birth between the mother and child is around 25%.[27] However, where combination antiretroviral drug treatment and Cesarian section are available, this risk can be reduced to as low as one percent.[27] Postnatal mother-to-child transmission may be largely prevented by complete avoidance of breast feeding; however, this has significant associated morbidity. Exclusive breast feeding and the provision of extended antiretroviral prophylaxis to the infant are also efficacious in avoiding transmission.[47]
Multiple infection
Unlike some other viruses, infection with HIV does not provide immunity against additional infections, particularly in the case of more genetically distant viruses. Both inter- and intra-clade multiple infections have been reported,[48] and even associated with more rapid disease progression.[49] Multiple infections are divided into two categories depending on the timing of the acquisition of the second strain. Coinfection refers to two strains that appear to have been acquired at the same time (or too close to distinguish). Reinfection (or superinfection) is infection with a second strain at a measurable time after the first. Both forms of dual infection have been reported for HIV in both acute and chronic infection around the world.[50][51][52][53]
Structure and genome

HIV is different in structure from other retroviruses. It is roughly spherical[54] with a diameter of about 120 nm, around 60 times smaller than a red blood cell, yet large for a virus.[55] It is composed of two copies of positive single-stranded RNA that codes for the virus's nine genes enclosed by a conical capsid composed of 2,000 copies of the viral protein p24.[56] The single-stranded RNA is tightly bound to nucleocapsid proteins, p7 and enzymes needed for the development of the virion such as reverse transcriptase, proteases, ribonuclease and integrase. A matrix composed of the viral protein p17 surrounds the capsid ensuring the integrity of the virion particle.[56] This is, in turn, surrounded by the viral envelope that is composed of two layers of fatty molecules called phospholipids taken from the membrane of a human cell when a newly formed virus particle buds from the cell. Embedded in the viral envelope are proteins from the host cell and about 70 copies of a complex HIV protein that protrudes through the surface of the virus particle.[56] This protein, known as Env, consists of a cap made of three molecules called glycoprotein (gp) 120, and a stem consisting of three gp41 molecules that anchor the structure into the viral envelope.[57] This glycoprotein complex enables the virus to attach to and fuse with target cells to initiate the infectious cycle.[57] Both these surface proteins, especially gp120, have been considered as targets of future treatments or vaccines against HIV.[58]
The RNA genome consists of at least seven structural landmarks (LTR, TAR, RRE, PE, SLIP, CRS, and INS) and nine genes (gag, pol, and env, tat, rev, nef, vif, vpr, vpu, and tev) encoding 19 proteins. Three of these genes, gag, pol, and env, contain information needed to make the structural proteins for new virus particles.[56] For example, env codes for a protein called gp160 that is broken down by a viral enzyme to form gp120 and gp41. The six remaining genes, tat, rev, nef, vif, vpr, and vpu (or vpx in the case of HIV-2), are regulatory genes for proteins that control the ability of HIV to infect cells, produce new copies of virus (replicate), or cause disease.[56] The two Tat proteins (p16 and p14) are transcriptional transactivators for the LTR promoter acting by binding the TAR RNA element. The TAR may also be processed into microRNAs that regulate the apoptosis genes ERCC1 and IER3.[59][60] The Rev protein (p19) is involved in shuttling RNAs from the nucleus and the cytoplasm by binding to the RRE RNA element. The Vif protein (p23) prevents the action of APOBEC3G (a cell protein that deaminates DNA:RNA hybrids and/or interferes with the Pol protein). The Vpr protein (p14) arrests cell division at G2/M. The Nef protein (p27) down-regulates CD4 (the major viral receptor), as well as the MHC class I and class II molecules.[61][62][63] Nef also interacts with SH3 domains. The Vpu protein (p16) influences the release of new virus particles from infected cells.[56] The ends of each strand of HIV RNA contain an RNA sequence called the long terminal repeat (LTR). Regions in the LTR act as switches to control production of new viruses and can be triggered by proteins from either HIV or the host cell. The Psi element is involved in viral genome packaging and recognized by Gag and Rev proteins. The SLIP element (TTTTTT) is involved in the frameshift in the Gag-Pol reading frame required to make functional Pol.[56]
Tropism
The term viral tropism refers to which cell types HIV infects. HIV can infect a variety of immune cells such as CD4+ T cells, macrophages, and microglial cells. HIV-1 entry to macrophages and CD4+ T cells is mediated through interaction of the virion envelope glycoproteins (gp120) with the CD4 molecule on the target cells and also with chemokine coreceptors.[57]
Macrophage (M-tropic) strains of HIV-1, or non-syncitia-inducing strains (NSI) use the β-chemokine receptor CCR5 for entry and are thus able to replicate in macrophages and CD4+ T cells.[64] This CCR5 coreceptor is used by almost all primary HIV-1 isolates regardless of viral genetic subtype. Indeed, macrophages play a key role in several critical aspects of HIV infection. They appear to be the first cells infected by HIV and perhaps the source of HIV production when CD4+ cells become depleted in the patient. Macrophages and microglial cells are the cells infected by HIV in the central nervous system. In tonsils and adenoids of HIV-infected patients, macrophages fuse into multinucleated giant cells that produce huge amounts of virus.
T-tropic isolates, or syncitia-inducing (SI) strains replicate in primary CD4+ T cells as well as in macrophages and use the α-chemokine receptor, CXCR4, for entry.[64][65][66] Dual-tropic HIV-1 strains are thought to be transitional strains of the HIV-1 virus and thus are able to use both CCR5 and CXCR4 as co-receptors for viral entry.
The α-chemokine SDF-1, a ligand for CXCR4, suppresses replication of T-tropic HIV-1 isolates. It does this by down-regulating the expression of CXCR4 on the surface of these cells. HIV that use only the CCR5 receptor are termed R5; those that only use CXCR4 are termed X4, and those that use both, X4R5. However, the use of coreceptor alone does not explain viral tropism, as not all R5 viruses are able to use CCR5 on macrophages for a productive infection[64] and HIV can also infect a subtype of myeloid dendritic cells,[67] which probably constitute a reservoir that maintains infection when CD4+ T cell numbers have declined to extremely low levels.
Some people are resistant to certain strains of HIV.[68] For example people with the CCR5-Δ32 mutation are resistant to infection with R5 virus as the mutation stops HIV from binding to this coreceptor, reducing its ability to infect target cells.
Sexual intercourse is the major mode of HIV transmission. Both X4 and R5 HIV are present in the seminal fluid, which is passed from a male to his sexual partner. The virions can then infect numerous cellular targets and disseminate into the whole organism. However, a selection process leads to a predominant transmission of the R5 virus through this pathway.[69][70][71] How this selective process works is still under investigation, but one model is that spermatozoa may selectively carry R5 HIV as they possess both CCR3 and CCR5 but not CXCR4 on their surface[72] and that genital epithelial cells preferentially sequester X4 virus.[73] In patients infected with subtype B HIV-1, there is often a co-receptor switch in late-stage disease and T-tropic variants appear that can infect a variety of T cells through CXCR4.[74] These variants then replicate more aggressively with heightened virulence that causes rapid T cell depletion, immune system collapse, and opportunistic infections that mark the advent of AIDS.[75] Thus, during the course of infection, viral adaptation to the use of CXCR4 instead of CCR5 may be a key step in the progression to AIDS. A number of studies with subtype B-infected individuals have determined that between 40 and 50% of AIDS patients can harbour viruses of the SI, and presumably the X4, phenotype.[76][77]
HIV-2 is much less pathogenic than HIV-1 and is restricted in its worldwide distribution. The adoption of "accessory genes" by HIV-2 and its more promiscuous pattern of coreceptor usage (including CD4-independence) may assist the virus in its adaptation to avoid innate restriction factors present in host cells. Adaptation to use normal cellular machinery to enable transmission and productive infection has also aided the establishment of HIV-2 replication in humans. A survival strategy for any infectious agent is not to kill its host but ultimately become a commensal organism. Having achieved a low pathogenicity, over time, variants more successful at transmission will be selected.[78]
[edit] Replication cycle

Entry to the cell
HIV enters macrophages and CD4+ T cells by the adsorption of glycoproteins on its surface to receptors on the target cell followed by fusion of the viral envelope with the cell membrane and the release of the HIV capsid into the cell.[79][80]
Entry to the cell begins through interaction of the trimeric envelope complex (gp160 spike) and both CD4 and a chemokine receptor (generally either CCR5 or CXCR4, but others are known to interact) on the cell surface.[79][80] gp120 binds to integrin α4β7 activating LFA-1 the central integrin involved in the establishment of virological synapses, which facilitate efficient cell-to-cell spreading of HIV-1.[81] The gp160 spike contains binding domains for both CD4 and chemokine receptors.[79][80] The first step in fusion involves the high-affinity attachment of the CD4 binding domains of gp120 to CD4. Once gp120 is bound with the CD4 protein, the envelope complex undergoes a structural change, exposing the chemokine binding domains of gp120 and allowing them to interact with the target chemokine receptor.[79][80] This allows for a more stable two-pronged attachment, which allows the N-terminal fusion peptide gp41 to penetrate the cell membrane.[79][80] Repeat sequences in gp41, HR1 and HR2 then interact, causing the collapse of the extracellular portion of gp41 into a hairpin. This loop structure brings the virus and cell membranes close together, allowing fusion of the membranes and subsequent entry of the viral capsid.[79][80]
After HIV has bound to the target cell, the HIV RNA and various enzymes, including reverse transcriptase, integrase, ribonuclease, and protease, are injected into the cell.[79] During the microtubule based transport to the nucleus, the viral single strand RNA genome is transcribed into double strand DNA, which is then integrated into a host chromosome.
HIV can infect dendritic cells (DCs) by this CD4-CCR5 route, but another route using mannose-specific C-type lectin receptors such as DC-SIGN can also be used.[82] DCs are one of the first cells encountered by the virus during sexual transmission. They are currently thought to play an important role by transmitting HIV to T-cells when the virus is captured in the mucosa by DCs.[82] The presence of FEZ-1, which occurs naturally in neurons, is believed to prevent the infection of cells by HIV.[83]
Replication and transcription
Shortly after the viral capsid enters the cell, an enzyme called reverse transcriptase liberates the single-stranded (+)RNA genome from the attached viral proteins and copies it into a complementary DNA (cDNA) molecule.[84] The process of reverse transcription is extremely error-prone, and the resulting mutations may cause drug resistance or allow the virus to evade the body's immune system. The reverse transcriptase also has ribonuclease activity that degrades the viral RNA during the synthesis of cDNA, as well as DNA-dependent DNA polymerase activity that creates a sense DNA from the antisense cDNA.[85] Together, the cDNA and its complement form a double-stranded viral DNA that is then transported into the cell nucleus. The integration of the viral DNA into the host cell's genome is carried out by another viral enzyme called integrase.[84]
This integrated viral DNA may then lie dormant, in the latent stage of HIV infection.[84] To actively produce the virus, certain cellular transcription factors need to be present, the most important of which is NF-κB (NF kappa B), which is upregulated when T-cells become activated.[86] This means that those cells most likely to be killed by HIV are those currently fighting infection.
During viral replication, the integrated DNA provirus is transcribed into mRNA, which is then spliced into smaller pieces. These small pieces are exported from the nucleus into the cytoplasm, where they are translated into the regulatory proteins Tat (which encourages new virus production) and Rev. As the newly produced Rev protein accumulates in the nucleus, it binds to viral mRNAs and allows unspliced RNAs to leave the nucleus, where they are otherwise retained until spliced.[87] At this stage, the structural proteins Gag and Env are produced from the full-length mRNA. The full-length RNA is actually the virus genome; it binds to the Gag protein and is packaged into new virus particles.
HIV-1 and HIV-2 appear to package their RNA differently; HIV-1 will bind to any appropriate RNA, whereas HIV-2 will preferentially bind to the mRNA that was used to create the Gag protein itself. This may mean that HIV-1 is better able to mutate (HIV-1 infection progresses to AIDS faster than HIV-2 infection and is responsible for the majority of global infections).
Assembly and release
The final step of the viral cycle, assembly of new HIV-1 virons, begins at the plasma membrane of the host cell. The Env polyprotein (gp160) goes through the endoplasmic reticulum and is transported to the Golgi complex where it is cleaved by protease and processed into the two HIV envelope glycoproteins gp41 and gp120. These are transported to the plasma membrane of the host cell where gp41 anchors the gp120 to the membrane of the infected cell. The Gag (p55) and Gag-Pol (p160) polyproteins also associate with the inner surface of the plasma membrane along with the HIV genomic RNA as the forming virion begins to bud from the host cell. Maturation either occurs in the forming bud or in the immature virion after it buds from the host cell. During maturation, HIV proteases cleave the polyproteins into individual functional HIV proteins and enzymes. The various structural components then assemble to produce a mature HIV virion.[88] This cleavage step can be inhibited by protease inhibitors. The mature virus is then able to infect another cell.
[edit] Genetic variability

HIV differs from many viruses in that it has very high genetic variability. This diversity is a result of its fast replication cycle, with the generation of 109 to 1010 virions every day, coupled with a high mutation rate of approximately 3 x 10−5 per nucleotide base per cycle of replication and recombinogenic properties of reverse transcriptase.[89][90]
This complex scenario leads to the generation of many variants of HIV in a single infected patient in the course of one day.[89] This variability is compounded when a single cell is simultaneously infected by two or more different strains of HIV. When simultaneous infection occurs, the genome of progeny virions may be composed of RNA strands from two different strains. This hybrid virion then infects a new cell where it undergoes replication. As this happens, the reverse transcriptase, by jumping back and forth between the two different RNA templates, will generate a newly synthesized retroviral DNA sequence that is a recombinant between the two parental genomes.[89] This recombination is most obvious when it occurs between subtypes.[89]
The closely related simian immunodeficiency virus (SIV) has evolved into many strains, classified by the natural host species. SIV strains of the African green monkey (SIVagm) and sooty mangabey (SIVsmm) are thought to have a long evolutionary history with their hosts. These hosts have adapted to the presence of the virus,[91] which is present at high levels in the host's blood but evokes only a mild immune response,[92] does not cause the development of simian AIDS,[93] and does not undergo the extensive mutation and recombination typical of HIV infection in humans.[94] In contrast, when these strains infect species that have not adapted to SIV ("heterologous" hosts such as rhesus or cynomologus macaques), the animals develop AIDS and the virus generates genetic diversity similar to what is seen in human HIV infection.[95] Chimpanzee SIV (SIVcpz), the closest genetic relative of HIV-1, is associated with increased mortality and AIDS-like symptoms in its natural host.[96] Both SIVcpz and HIV-1 appear to have been transmitted relatively recently to chimpanzee and human populations, so their hosts have not yet adapted to the virus.[91] Both viruses have also lost a function of the Nef gene that is present in most SIVs; without this function, T cell depletion is more likely, leading to immunodeficiency.[96]
Three groups of HIV-1 have been identified on the basis of differences in the envelope (env) region: M, N, and O.[97] Group M is the most prevalent and is subdivided into eight subtypes (or clades), based on the whole genome, which are geographically distinct.[98] The most prevalent are subtypes B (found mainly in North America and Europe), A and D (found mainly in Africa), and C (found mainly in Africa and Asia); these subtypes form branches in the phylogenetic tree representing the lineage of the M group of HIV-1. Coinfection with distinct subtypes gives rise to circulating recombinant forms (CRFs). In 2000, the last year in which an analysis of global subtype prevalence was made, 47.2% of infections worldwide were of subtype C, 26.7% were of subtype A/CRF02_AG, 12.3% were of subtype B, 5.3% were of subtype D, 3.2% were of CRF_AE, and the remaining 5.3% were composed of other subtypes and CRFs.[99] Most HIV-1 research is focused on subtype B; few laboratories focus on the other subtypes.[100] The existence of a fourth group, "P", has been hypothesised based on a virus isolated in 2009.[101][102][103] The strain is apparently derived from gorilla SIV (SIVgor), first isolated from western lowland gorillas in 2006.[101]
The genetic sequence of HIV-2 is only partially homologous to HIV-1 and more closely resembles that of SIVsmm.
Diagnosis
Many HIV-positive people are unaware that they are infected with the virus.[104] For example, less than 1% of the sexually active urban population in Africa have been tested and this proportion is even lower in rural populations.[104] Furthermore, only 0.5% of pregnant women attending urban health facilities are counselled, tested or receive their test results.[104] Again, this proportion is even lower in rural health facilities.[104] Since donors may therefore be unaware of their infection, donor blood and blood products used in medicine and medical research are routinely screened for HIV.[105]
HIV-1 testing consists of initial screening with an enzyme-linked immunosorbent assay (ELISA) to detect antibodies to HIV-1. Specimens with a nonreactive result from the initial ELISA are considered HIV-negative unless new exposure to an infected partner or partner of unknown HIV status has occurred. Specimens with a reactive ELISA result are retested in duplicate.[106] If the result of either duplicate test is reactive, the specimen is reported as repeatedly reactive and undergoes confirmatory testing with a more specific supplemental test (e.g., Western blot or, less commonly, an immunofluorescence assay (IFA)). Only specimens that are repeatedly reactive by ELISA and positive by IFA or reactive by Western blot are considered HIV-positive and indicative of HIV infection. Specimens that are repeatedly ELISA-reactive occasionally provide an indeterminate Western blot result, which may be either an incomplete antibody response to HIV in an infected person, or nonspecific reactions in an uninfected person.[107] Although IFA can be used to confirm infection in these ambiguous cases, this assay is not widely used. Generally, a second specimen should be collected more than a month later and retested for persons with indeterminate Western blot results. Although much less commonly available, nucleic acid testing (e.g., viral RNA or proviral DNA amplification method) can also help diagnosis in certain situations.[106] In addition, a few tested specimens might provide inconclusive results because of a low quantity specimen. In these situations, a second specimen is collected and tested for HIV infection.
Modern HIV testing is extremely accurate. The chance of a false-positive result in the two-step testing protocol is estimated to be 0.0004% to 0.0007% in the general U.S. population.[108][109][110][111]
Treatment
- See also Antiretroviral drug
There is currently no publicly available vaccine or cure for HIV or AIDS.[112][113] However, a vaccine that is a combination of two previously unsuccessful vaccine candidates was reported in September 2009 to have resulted in a 30% reduction in infections in a trial conducted in Thailand.[114] Additionally, a course of antiretroviral treatment administered immediately after exposure, referred to as post-exposure prophylaxis, is believed to reduce the risk of infection if begun as quickly as possible.[115] However, due to the incomplete protection provided by the vaccine and/or post-exposure prophylaxis, the avoidance of exposure to the virus is expected to remain the only reliable way to escape infection for some time yet. Current treatment for HIV infection consists of highly active antiretroviral therapy, or HAART.[116] This has been highly beneficial to many HIV-infected individuals since its introduction in 1996, when the protease inhibitor-based HAART initially became available.[117] Current HAART options are combinations (or "cocktails") consisting of at least three drugs belonging to at least two types, or "classes," of antiretroviral agents. Typically, these classes are two nucleoside analogue reverse transcriptase inhibitors (NARTIs or NRTIs) plus either a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor (NNRTI). New classes of drugs such as entry inhibitors provide treatment options for patients who are infected with viruses already resistant to common therapies, although they are not widely available and not typically accessible in resource-limited settings. Because AIDS progression in children is more rapid and less predictable than in adults, particularly in young infants, more aggressive treatment is recommended for children than adults.[118] In developed countries where HAART is available, doctors assess their patients thoroughly: measuring the viral load, how fast CD4 declines, and patient readiness. They then decide when to recommend starting treatment.[119]
HAART neither cures the patient nor does it uniformly remove all symptoms; high levels of HIV-1, often HAART resistant, return if treatment is stopped.[120][121] Moreover, it would take more than a lifetime for HIV infection to be cleared using HAART.[122] Despite this, many HIV-infected individuals have experienced remarkable improvements in their general health and quality of life, which has led to a large reduction in HIV-associated morbidity and mortality in the developed world.[117][123][124] One study suggests the average life expectancy of an HIV infected individual is 32 years from the time of infection if treatment is started when the CD4 count is 350/µL.[125] In the absence of HAART, progression from HIV infection to AIDS has been observed to occur at a median of between nine to ten years and the median survival time after developing AIDS is only 9.2 months.[11] However, HAART sometimes achieves far less than optimal results, in some circumstances being effective in less than fifty percent of patients. This is due to a variety of reasons such as medication intolerance/side effects, prior ineffective antiretroviral therapy and infection with a drug-resistant strain of HIV. However, non-adherence and non-persistence with antiretroviral therapy is the major reason most individuals fail to benefit from HAART.[126] The reasons for non-adherence and non-persistence with HAART are varied and overlapping. Major psychosocial issues, such as poor access to medical care, inadequate social supports, psychiatric disease and drug abuse contribute to non-adherence. The complexity of these HAART regimens, whether due to pill number, dosing frequency, meal restrictions or other issues along with side effects that create intentional non-adherence also contribute to this problem.[127][128][129] The side effects include lipodystrophy, dyslipidemia, insulin resistance, an increase in cardiovascular risks, and birth defects.[130][131]
The timing for starting HIV treatment is still debated. There is no question that treatment should be started before the patient's CD4 count falls below 200, and most national guidelines say to start treatment once the CD4 count falls below 350; but there is some evidence from cohort studies that treatment should be started before the CD4 count falls below 350.[123][132] In those countries where CD4 counts are not available, patients with WHO stage III or IV disease[133] should be offered treatment.
Anti-retroviral drugs are expensive, and the majority of the world's infected individuals do not have access to medications and treatments for HIV and AIDS.[134] Research to improve current treatments includes decreasing side effects of current drugs, further simplifying drug regimens to improve adherence, and determining the best sequence of regimens to manage drug resistance. Unfortunately, only a vaccine is thought to be able to halt the pandemic. This is because a vaccine would cost less, thus being affordable for developing countries, and would not require daily treatment.[134] However, after over 20 years of research, HIV-1 remains a difficult target for a vaccine.[134]
Treatments in development
Media reports in 2008 and a publication in the New England Journal of Medicine in 2009 described the anecdotal case of an HIV-positive patient of a Berlin doctor, Gero Hütter. The patient, who had both acute myelogenous leukemia (AML) and HIV infection, was said by some to be "functionally cured" of his HIV following a bone marrow transplant for AML. The bone marrow donor had been selected as homozygous for a CCR5-Δ32 mutation (which confers resistance to "almost all strains of HIV").[135][136] After 600 days without antiretroviral drug treatment, HIV levels in the patient's blood, bone marrow and bowel were below the limit of detection, although the authors note that the virus is likely present in other tissues. Researchers cautioned that it would be premature to consider this treatment a possible cure because of its anecdotal nature, the mortality risk associated with bone marrow transplants and other concerns.[137][138]
HIV latent reservoir
Despite the success of highly active antiretroviral therapy (HAART) in controlling HIV infection and reducing HIV-associated mortality, current drug regimens are unable to completely eradicate HIV infection. Many people on HAART achieve suppression of HIV to levels below the limit of detection of standard clinical assays for many years. However, upon withdrawal of HAART, HIV viral loads rebound quickly with a concomitant decline in CD4+ T-Cells, which, in most cases, absent a resumption of treatment, leads to AIDS.
To successfully reproduce itself, HIV must convert its RNA genome to DNA, which is then imported into the host cell's nucleus and inserted into the host genome through the action of HIV integrase. Because HIV's primary cellular target, CD4+ T-Cells, function as the memory cells of the immune system, integrated HIV can remain dormant for the duration of these cell's lifetime. Memory T-Cells may survive for many years and possibly for decades. The latent HIV reservoir can be measured by co-culturing CD4+ T-Cells from infected patients with CD4+ T-Cells from uninfected donors and measuring HIV protein or RNA.[139]
The failure of vaccine candidates to protect against HIV infection and progression to AIDS has led to a renewed focus on the biological mechanisms responsible for HIV latency. A limited period of therapy combining anti-retrovirals with drugs targeting the latent reservoir may one day allow for total eradication of HIV infection.[140]
Prognosis
Without treatment, the net median survival time after infection with HIV is estimated to be 9 to 11 years, depending on the HIV subtype,[141] and the median survival rate after diagnosis of AIDS in resource-limited settings where treatment is not available ranges between 6 and 19 months, depending on the study.[142] In areas where it is widely available, the development of HAART as effective therapy for HIV infection and AIDS reduced the death rate from this disease by 80%, and raised the life expectancy for a newly diagnosed HIV-infected person to 20–50 years.[143][144]
As new treatments continue to be developed and because HIV continues to evolve resistance to treatments, estimates of survival time are likely to continue to change. Without antiretroviral therapy, death normally occurs within a year after the individual progresses to AIDS.[11] Most patients die from opportunistic infections or malignancies associated with the progressive failure of the immune system.[7] The rate of clinical disease progression varies widely between individuals and has been shown to be affected by many factors such as host susceptibility and immune function[68][145][146] health care and co-infections,[7][11] as well as which particular strain of the virus is involved.[147][148][149]
Epidemiology


UNAIDS and the WHO estimate that AIDS has killed more than 25 million people since it was first recognized in 1981, making it one of the most destructive pandemics in recorded history. Despite recent improved access to antiretroviral treatment and care in many regions of the world, the AIDS pandemic claimed an estimated 2.8 million (between 2.4 and 3.3 million) lives in 2005 of which more than half a million (570,000) were children.[3]
In 2007, between 30.6 and 36.1 million people were believed to live with HIV, and it killed an estimated 2.1 million people that year, including 330,000 children; there were 2.5 million new infections.[141]
Sub-Saharan Africa remains by far the worst-affected region, with an estimated 21.6 to 27.4 million people currently living with HIV. Two million (1.5–3.0 million) of them are children younger than 15 years of age. More than 64% of all people living with HIV are in sub-Saharan Africa, as are more than three quarters of all women living with HIV. In 2005, there were 12.0 million (10.6–13.6 million) AIDS orphans living in sub-Saharan Africa 2005.[3]
South & South East Asia are second-worst affected with 15% of the total. AIDS accounts for the deaths of 500,000 children in this region. South Africa has the largest number of HIV patients in the world followed by Nigeria.[150] India has an estimated 2.5 million infections (0.23% of population), making India the country with the third largest population of HIV patients. In the 35 African nations with the highest prevalence, average life expectancy is 48.3 years—6.5 years less than it would be without the disease.[151]
The latest evaluation report of the World Bank's Operations Evaluation Department assesses the development effectiveness of the World Bank's country-level HIV/AIDS assistance defined as policy dialogue, analytic work, and lending with the explicit objective of reducing the scope or impact of the AIDS epidemic.[152] This is the first comprehensive evaluation of the World Bank's HIV/AIDS support to countries, from the beginning of the epidemic through mid-2004. Because the Bank aims to assist in implementation of national government programmes, their experience provides important insights on how national AIDS programmes can be made more effective.
The development of HAART as effective therapy for HIV infection has substantially reduced the death rate from this disease in those areas where these drugs are widely available.[117] As the life expectancy of persons with HIV has increased in countries where HAART is widely used, the continuing spread of the disease has caused the number of persons living with HIV to increase substantially.
In Africa, the number of MTCT and the prevalence of AIDS is beginning to reverse decades of steady progress in child survival. Countries such as Uganda are attempting to curb the MTCT epidemic by offering VCT (voluntary counselling and testing), PMTCT (prevention of mother-to-child transmission) and ANC (ante-natal care) services, which include the distribution of antiretroviral therapy.
History
Origins
- See History of known cases and spread for early cases of HIV / AIDS
HIV is thought to have originated in non-human primates in sub-Saharan Africa and was transferred to humans late in the 19th or early in the 20th century.[153] The first paper recognizing a pattern of opportunistic infections characteristic of AIDS was published in 1981.[154]
Both HIV-1 and HIV-2 are believed to have originated in West-Central Africa and to have jumped species (a process known as zoonosis) from non-human primates to humans. HIV-1 appears to have originated in southern Cameroon through the evolution of SIV(cpz), a simian immunodeficiency virus (SIV) that infects wild chimpanzees (Pan troglodytes troglodytes).[155][156] The closest relative of HIV-2 is SIV(agm), a virus of the sooty mangabey (Cercocebus atys), an Old World monkey of Guinea-Bissau, Gabon, and Cameroon.[17] New World monkeys such as the owl monkey are resistant to HIV-1 infection, possibly because of a genomic fusion of two viral resistance genes.[157]
Discovery
AIDS was first clinically observed between late 1980 and early 1981. A group of five men showed symptoms of Pneumocystis carinii pneumonia (PCP), a rare opportunistic infection that was known to present itself in people with very compromised immune systems. Soon thereafter, another set of men developed a rare skin cancer called Kaposi’s sarcoma (KP). Many more cases of PCP and KP quickly emerged, alerting U.S. Centers for Disease Control and Prevention (CDC). A CDC task force was formed to monitor the outbreak. After recognizing a pattern of anomalous symptoms presenting themselves in patients, the task force named the condition acquired immune deficiency syndrome (AIDS).[158]
In 1983, two separate research groups led by Robert Gallo and Luc Montagnier independently declared that a novel retrovirus may have been infecting AIDS patients, and published their findings in the same issue of the journal Science.[159][160] Gallo claimed that a virus his group had isolated from an AIDS patient was strikingly similar in shape to other human T-lymphotropic viruses (HTLVs) his group had been the first to isolate. Gallo's group called their newly isolated virus HTLV-III. At the same time, Montagnier's group isolated a virus from a patient presenting lymphadenopathy (swelling of the lymph nodes) of the neck and physical weakness, two classic symptoms of AIDS. Contradicting the report from Gallo's group, Montagnier and his colleagues showed that core proteins of this virus were immunologically different from those of HTLV-I. Montagnier's group named their isolated virus lymphadenopathy-associated virus (LAV).[158]
Whether Gallo or Montagnier deserve more credit for the discovery of the virus that causes AIDS has been a matter of considerable controversy. Together with his colleague Françoise Barré-Sinoussi, Montagnier was awarded one half of the 2008 Nobel Prize in Physiology or Medicine for his "discovery of human immunodeficiency virus".[161] Harald zur Hausen also shared the Prize for his discovery that human papilloma virus leads to cervical cancer, but Gallo was left out.[162] Gallo said that it was "a disappointment" that he was not named a co-recipient.[163] Montagnier said he was "surprised" Gallo was not recognized by the Nobel Committee: "It was important to prove that HIV was the cause of AIDS, and Gallo had a very important role in that. I'm very sorry for Robert Gallo."[162]
AIDS denialism
Some individuals, including some scientists who are not recognized experts on HIV,[164] question the connection between HIV and AIDS,[165] the existence of HIV itself, or the validity of HIV testing and treatment methods.[164][166] These claims, known as AIDS denialism, have been examined and rejected by the worldwide scientific community,[167] although they have had a political impact, particularly in South Africa, where the government's official promotion of AIDS denialism was responsible for its ineffective response to that country's AIDS epidemic.[168][169][170]
African swine fever virus
20.46

African swine fever virus (ASFV) is the causative agent of African swine fever. ASFV is a large double-stranded DNA virus which replicates in the cytoplasm of infected cells and is the only member of the Asfarviridae family.[1] ASFV infects domestic pigs, warthogs and bushpigs, as well as soft ticks (Ornithodoros),which are likely vectors. ASFV is the only virus with a DNA genome that is transmitted by arthropods. The virus causes a lethal haemorraghic disease in domestic pigs. Some isolates can cause death of animals within as little as a week after infection. In all other species, the virus causes no obvious disease. ASFV is endemic to Sub-Saharan Africa and exists in the wild through a cycle of infection between ticks and wild pigs/bushpigs/warthogs. ASFV was first described after European settlers brought pigs into areas endemic with ASFV and as such is an example of an 'emerging infection'.
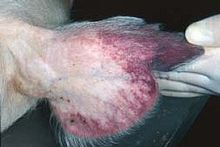
The clinical symptoms of ASFV are very similar to classical swine fever virus and the two diseases normally have to be distinguished by laboratory diagnosis.
African swine fever was restricted to the continent that bears its name until 1957 when the disease was reported in Lisbon, Portugal. A further outbreak occurred here in 1960. Subsequent to these initial introductions the disease became established in the Iberian peninsula and sporadic outbreaks occurred in France, Belgium and other European countries during the 1980s. Both Spain and Portugal had managed to eradicate the disease by the mid 1990s through a slaughter policy. During the late 1970s ASFV crossed the Atlantic Ocean and outbreaks were reported in some Caribbean islands, including Cuba and the Dominican Republic. Major outbreaks of ASFV in Africa are regularly reported to the World Organisation for Animal Health (previously called l'office international des épizooties (OIE)). The most recent outbreak of ASFV outside of Africa started at the beginning of 2007 in Georgia (country) and has since spread to the neighbouring countries of Armenia, Azerbaijan and Russia.

The appearance of ASF in the Western hemisphere at the same time as the emergence of AIDS led to some interest in whether the two were related and a report appeared in the Lancet supporting this in 1986. However, AIDS is now known to be caused by human immunodeficiency virus (HIV), no link between ASFV and AIDS has been proven or is suspected.
Virus structure and replication

ASFV is a large double-stranded DNA virus with a genome containing at least 150 genes. The number of genes differs slightly between different isolates of the virus. ASFV has similarities to the other large DNA viruses e.g., poxvirus, iridovirus, mimivirus. In common with other viral haemorrhagic fevers, the main target cells for replication are those of monocyte, macrophage lineage. The virus causes a haemorrhagic fever with high mortality rates in pigs, but persistently infects its natural hosts, warthogs, bushpigs and soft ticks of the Ornithodoros species with no disease signs. The virus encodes enzymes required for replication and transcription of the genome, including elements of a base excision repair system, structural proteins and many proteins that are not essential for replication in cells but have roles in virus survival and transmission in its hosts. Virus replication takes place in perinuclear factory areas. Assembly of the icosahedral capsid occurs on modified membranes from the endoplasmic reticulum. Products from proteolytically processed polyproteins form the core shell between the internal membrane and the nucleoprotein core. An additional outer membrane is gained as particles bud from the plasma membrane. The virus encodes proteins that inhibit signalling pathways in infected macrophages and thus modulate transcriptional activation of immune response genes. In addition the virus encodes proteins which inhibit apoptosis of infected cells to facilitate production of progeny virions. Viral membrane proteins with similarity to cellular adhesion proteins modulate interaction of virus-infected cells and extracellular virions with host components.[1]

